EnrichMap supports user defined gene weights#
This tutorial demonstrates how to use EnrichMap when the pathway of interest has gene weight information.
import os
os.environ["PYTHONWARNINGS"] = "ignore"
import warnings
warnings.filterwarnings("ignore")
Import required packages for minimal example.
import scanpy as sc
import enrichmap as em
sc.set_figure_params(frameon=False)
Read in the dataset:
adata = sc.read(
"adata_breast.h5ad",
backup_url="https://github.com/secrierlab/EnrichMap/raw/main/tests/dataset/adata_breast.h5ad",
)
100%|██████████| 69.1M/69.1M [03:33<00:00, 339kB/s]
As the data is shared as raw counts, we normalise them.
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
Here, we investigate EGFR pathway in breast cancer. The weighted gene signature is taken from Schubert et al (2018).
The gene_weights argument expects a nested dictionary, where the outer key is the signature name
(used for storing scores in adata.obs) and the inner dictionary maps gene names to their weights.
This structure is required even for a single signature, as it allows EnrichMap to handle multiple
weighted signatures in a single call — the same way gene_set accepts a dictionary of multiple gene lists.
# Structure:
gene_weights = {
"signature_name": {
"Gene1": weight1,
"Gene2": weight2,
...
}
}
nested_dict = {
"EGFR pathway": {
"DUSP6": 12.62931259,
"DUSP5": 7.943039507,
"FOSL1": 7.647829795,
"DUSP4": 6.967179671,
"PHLDA1": 6.619626129,
"EGR3": 6.271415501,
"EPHA2": 5.411184102,
"ODC1": 4.678550829,
"PNP": 4.643619942,
"SPOCD1": 4.295689401,
"TUBB4B": 4.084510208,
"LAMC2": 3.920115723,
"TRIB1": 3.917794383,
"PTHLH": 3.613976653,
"PHLDA2": 3.505138341,
"PLEK2": 3.439256964,
"AEN": 2.987402897,
"EGR4": 2.953076385,
"HAS3": 2.923737765,
"BZW1": 2.746151949,
"SH2D5": 2.432132976,
"DSPP": 2.350387113,
"SLC45A3": 1.902438902,
"SPATA2L": 1.756497668,
"MYEOV": 1.726632434,
"HMOX2": 1.719637973,
"VPS9D1-AS1": 1.601747009,
"KCNF1": 1.57587573,
"LOC101927363": 1.510406613,
"XRCC3": 1.487957075,
"DCAF13": 1.434232957,
"LOC105375839": 1.428616839,
"CFL1": 1.329245479,
"ADORA1": 1.267810008,
"PWP2": 1.172488384,
"MDFI": 1.160109073,
"GPR75": 1.037039515,
"TUBB7P": 0.99687917,
"MYO1A": 0.99477812,
"CBARP": 0.979442479,
"EXD3": 0.779924513,
"TRH": 0.771964661,
"CHRNA10": 0.6899809,
}
}
We now calculate EnrichMap scores for the EGFR pathway. As there is more than one slide in this dataset, we specify the batch information stored in adata.obs. Note that when using weighted signatures, we use the gene_weights argument instead of gene_set.
em.tl.score(
adata,
gene_weights=nested_dict,
batch_key="batch"
)
Scoring EGFR pathway: 34/43 genes found: 100%|██████████| 1/1 [00:00<00:00, 1.20it/s]
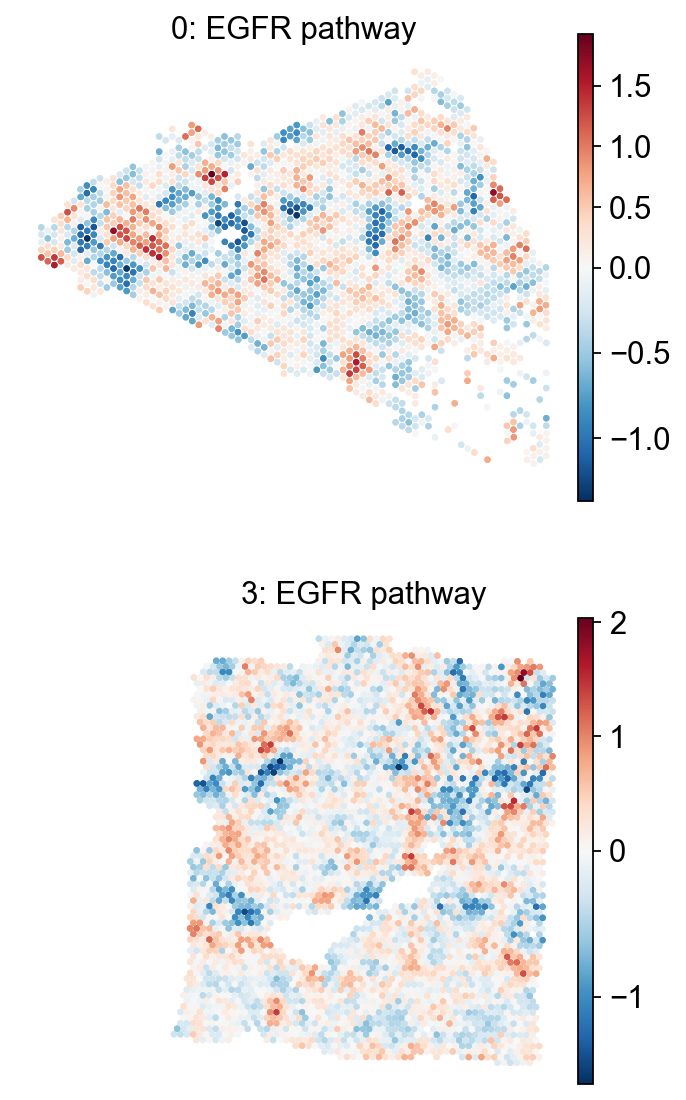
em.pl.spatial_enrichmap(
adata,
score_key=["EGFR pathway_score"],
size=2,
library_key="batch",
library_id=["0", "3"],
cmap="RdBu_r",
shape=None
)
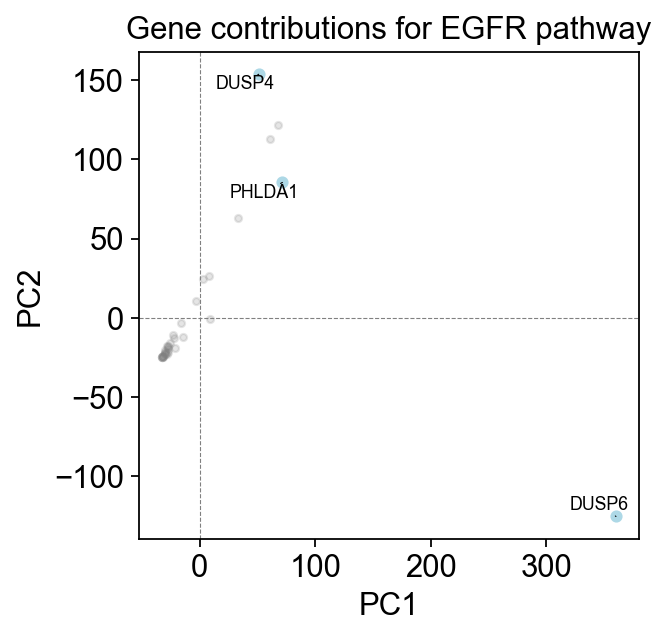
Let’s investigate which genes contributed most to the EGFR pathway score.
em.pl.gene_contributions_pca(
adata,
score_key="EGFR pathway",
top_n_genes=3
)